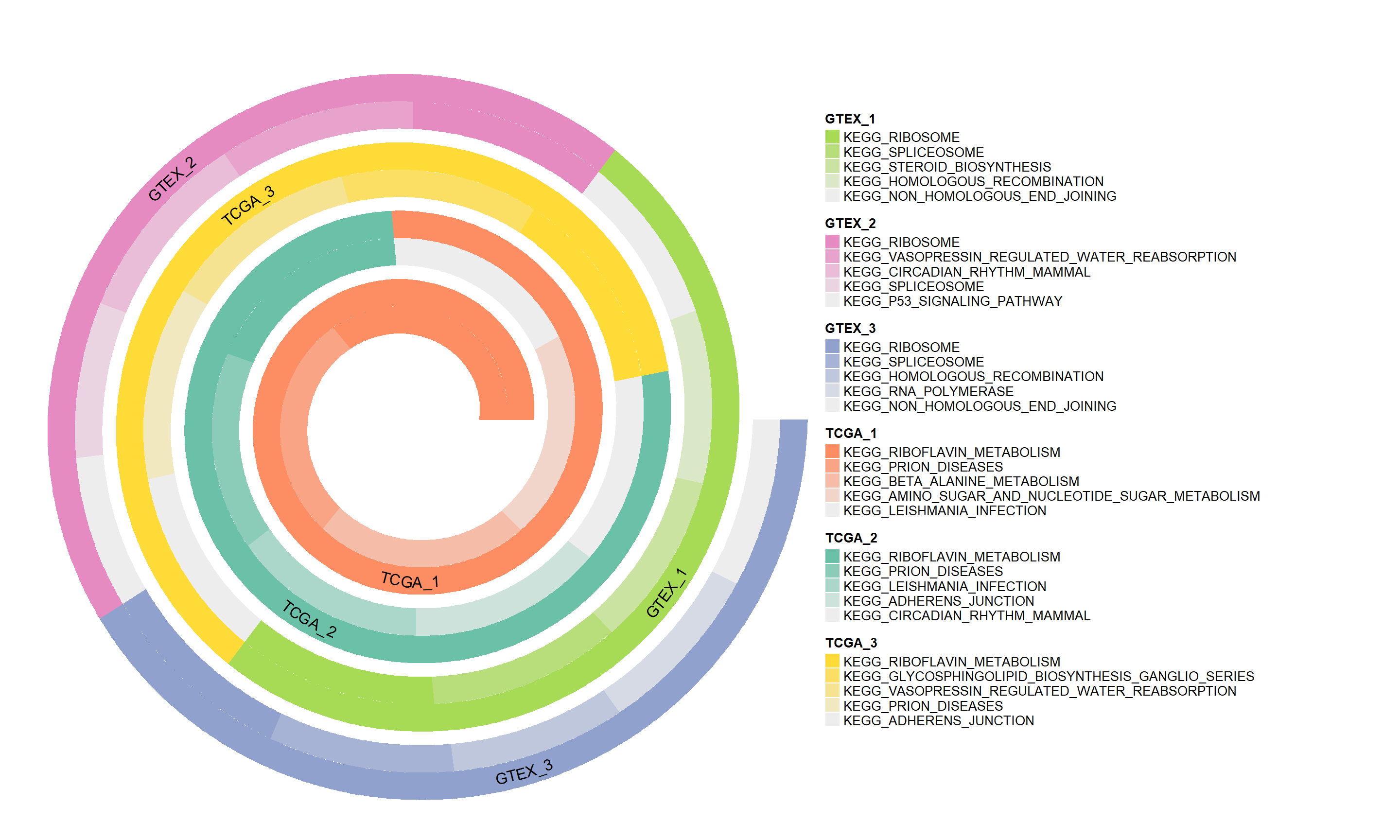
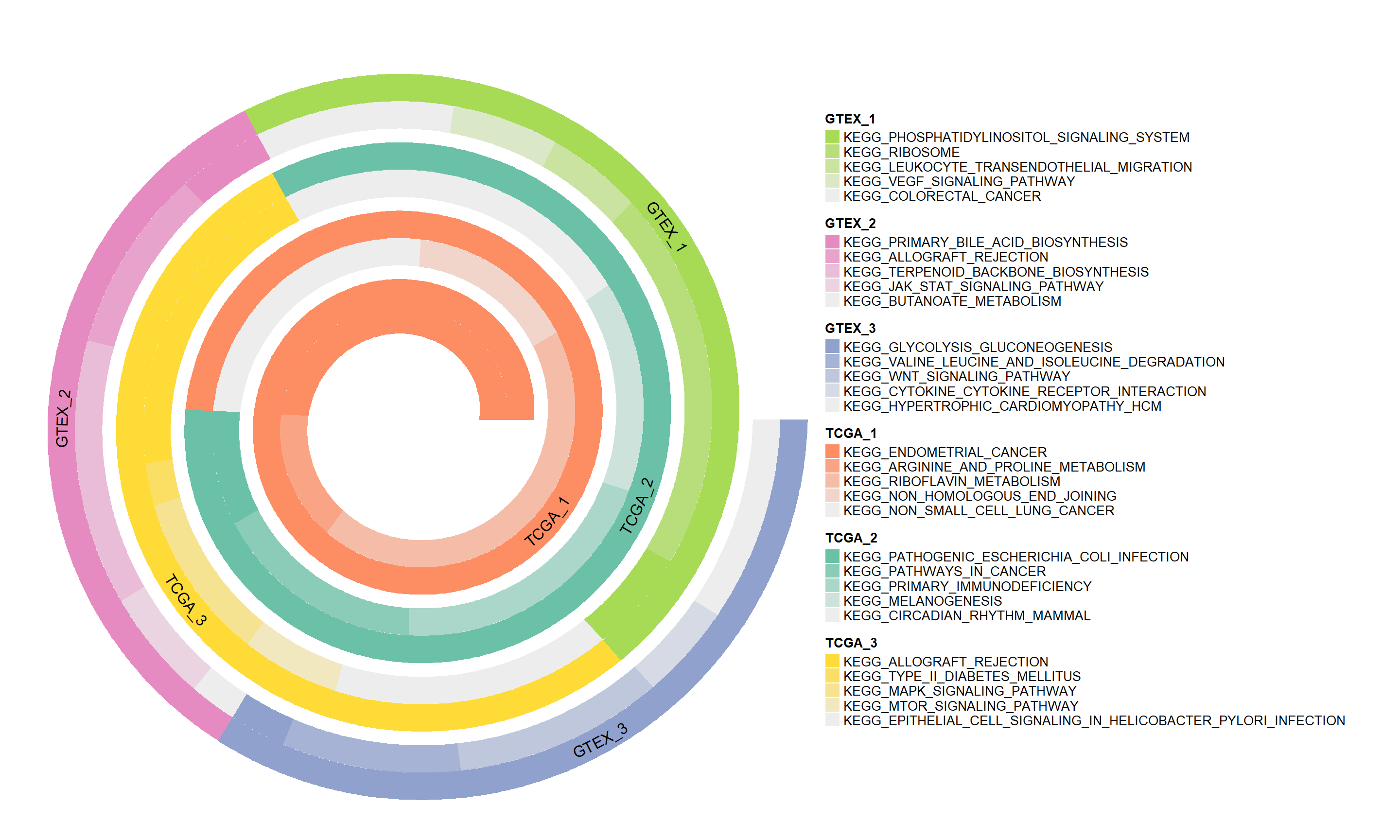
The spiral plots generated by this module provide an intuitive visual display, depicting the expression changes in different biological pathways across samples through gradient colors and spatial arrangement. This visualization method helps to more clearly identify and compare the activity differences in key pathways, thereby deepening the understanding of physiological mechanisms.
tumor <-readRDS("../test_TransProR/generated_data1/removebatch_SKCM_Skin_TCGA_exp_tumor.rds")normal <-readRDS('../test_TransProR/generated_data1/removebatch_SKCM_Skin_Normal_TCGA_GTEX_count.rds')# Merge the datasets, ensuring both have genes as row namesall_count_exp <-merge(tumor, normal, by ="row.names")all_count_exp <- tibble::column_to_rownames(all_count_exp, var ="Row.names") # Set the row names# Drawing data# all_count_exp <- log_transform(all_count_exp)DEG_deseq2 <-readRDS('../test_TransProR/Select DEGs/DEG_deseq2.Rdata')#head(all_count_exp, 1)head(DEG_deseq2, 5)
# Convert from SYMBOL to ENTREZID for convenient enrichment analysis later. It's crucial to do this now as a direct conversion may result in a reduced set of genes due to non-one-to-one correspondence.# DEG_deseq2# Retrieve gene listgene <-rownames(DEG_deseq2)# Perform conversiongene =bitr(gene, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
'select()' returned 1:many mapping between keys and columns
Warning in bitr(gene, fromType = "SYMBOL", toType = "ENTREZID", OrgDb =
"org.Hs.eg.db"): 43.37% of input gene IDs are fail to map...
# Remove duplicates and mergegene <- dplyr::distinct(gene, SYMBOL, .keep_all=TRUE)# Extract the SYMBOL column as a vector from the first datasetsymbols_vector <- gene$SYMBOL# Use the SYMBOL column to filter corresponding rows from the second dataset by row namesDEG_deseq2 <- DEG_deseq2[rownames(DEG_deseq2) %in% symbols_vector, ]head(DEG_deseq2, 5)
Diff_deseq2 <-filter_diff_genes( DEG_deseq2, p_val_col ="pvalue", log_fc_col ="log2FoldChange",p_val_threshold =0.05, log_fc_threshold =3)# First, obtain a list of gene names from the row names of the first datasetgene_names <-rownames(Diff_deseq2)# Find the matching rows in the second dataframematched_rows <- all_count_exp[gene_names, ]# Calculate the mean for each rowaverages <-rowMeans(matched_rows, na.rm =TRUE)# Append the averages as a new column to the first dataframeDiff_deseq2$average <- averagesDiff_deseq2$ID <-rownames(Diff_deseq2)Diff_deseq2$changetype <-ifelse(Diff_deseq2$change =='up', 1, -1)# Define a small threshold valuesmall_value <- .Machine$double.xmin# Before calculating -log10, replace zeroes with the small threshold value and assign it to a new columnDiff_deseq2$log_pvalue <-ifelse(Diff_deseq2$pvalue ==0, -log10(small_value), -log10(Diff_deseq2$pvalue))# Extract the expression data corresponding to the differentially expressed genesheatdata_deseq2 <- all_count_exp[rownames(Diff_deseq2), ]set.seed(123)# Preparing heatdata for visualizationHeatdataDeseq2 <- TransProR::process_heatdata(heatdata_deseq2, selection =2, custom_names =NULL, num_names_per_group =3, prefix_length =4)HeatdataDeseq2 <-as.matrix(HeatdataDeseq2)
## Using the msigdbr package to download and prepare for GSVA analysis with KEGG and GO gene sets## KEGGKEGG_df_all <-msigdbr(species ="Homo sapiens", # Homo sapiens or Mus musculuscategory ="C2",subcategory ="CP:KEGG") KEGG_df <- dplyr::select(KEGG_df_all, gs_name, gs_exact_source, gene_symbol)kegg_list <-split(KEGG_df$gene_symbol, KEGG_df$gs_name) # Grouping gene symbols by gs_namehead(kegg_list)
results1$PathwayColor <- Pathwaycolor2results2$PathwayColor <- Pathwaycolor2# Create a color mapping for the Sample columnSample_colors2 <-setNames(Samplecolor2, unique(results1$Sample))Sample_colors2 <-setNames(Samplecolor2, unique(results2$Sample))# Add SampleColor column to the DataFrameresults1$SampleColor <- Sample_colors2[results1$Sample]results2$SampleColor <- Sample_colors2[results2$Sample]# View the resultsprint(head(results1))